
Calculators & Predictors
Execute high quality physico-chemical calculations and predictions.
Summary
Virtual property predictions for tangible advancements
Our industry leading solution for property predictions offers a wide range of quick chemical calculations for multiple endpoints, combining outstanding accuracy, great availability, consistency and integration options. From easy-to-use plugins to fully customizable command line tools, the Calculators & Predictors are available via all main Chemaxon products. Extensive API and training options provide further possibilities for integration and customization.

Try it free

Download
Features
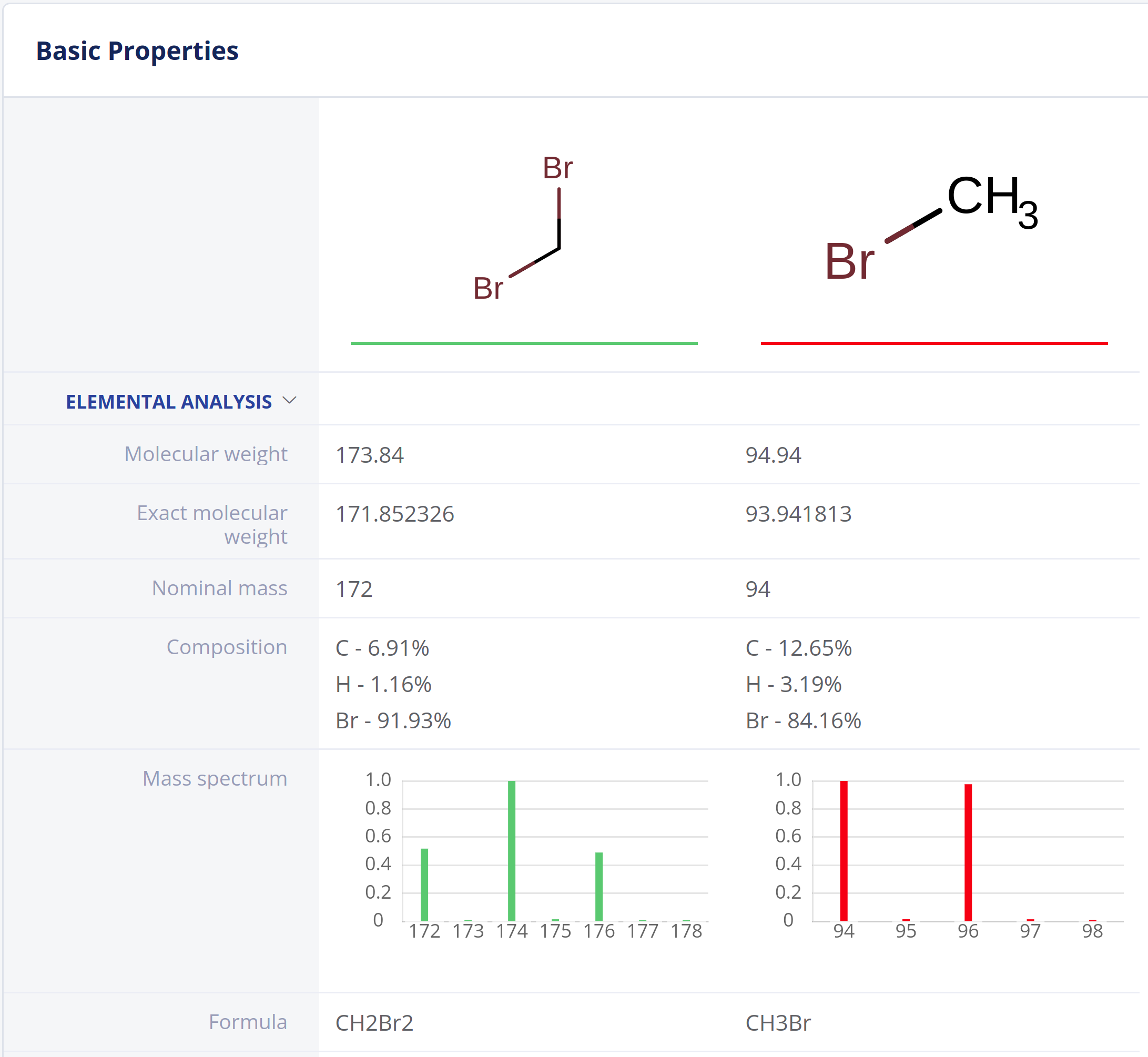
Elemental Analysis
Elemental Analysis provides fundamental molecular properties for general chemistry workflows and analytical scientists.
- Exact and nominal mass, molecular weight and atom count
- Elemental composition
- Molecular formulae
- Molecular composition
- Mass spectrum (isotope distribution) calculation.
Features
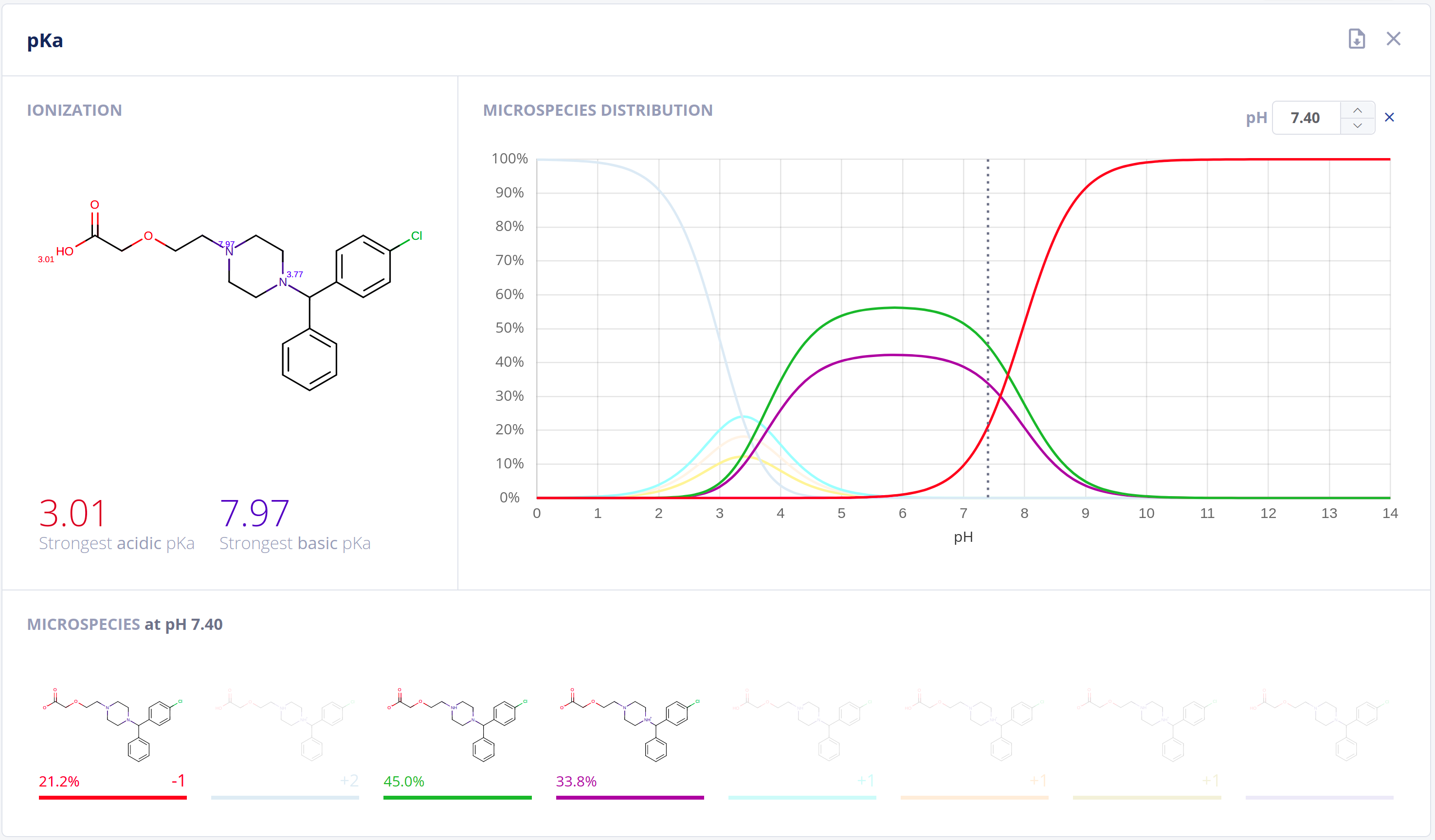
Protonation
Ionization has an essential impact on compound behavior in nearly all protic environments. On top of its application in general and analytical chemistry, relationships to drug pharmacokinetics and pharmacodynamics are well-established. Acidic and basic dissociation constants (pKa) and the microspecies distribution at different pH values are important to quantitatively describe ionization and comprehend physical and biological processes.The Protonation bundle includes:
- Highly-accurate calculation of pKa values along with pH dependent distribution plots of relevant microspecies in water - pKa documentation
- Calculation of the major microspecies form at a given pH value - Major microspecies documentation
- Isoelectric point calculation
Features
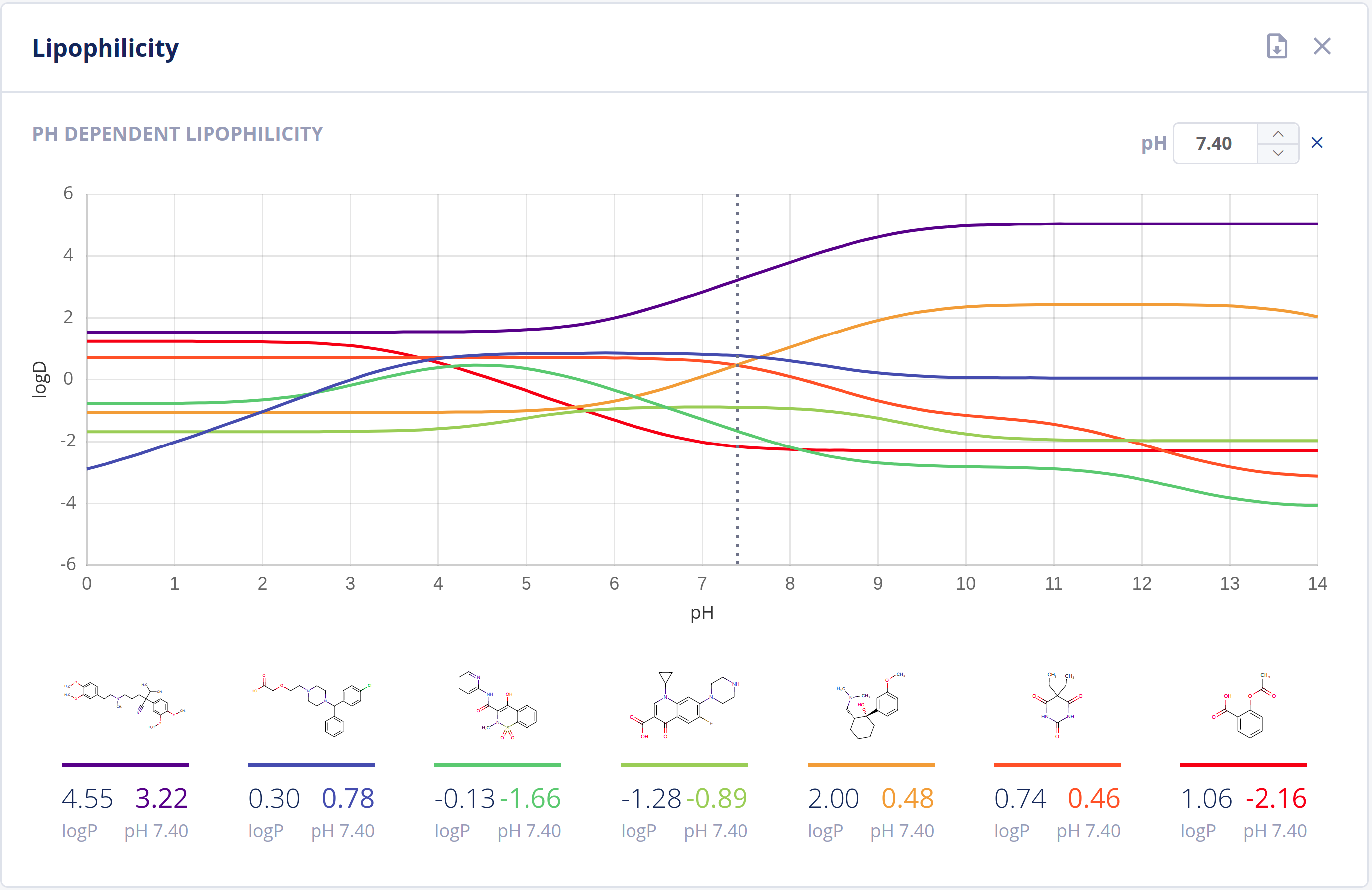
Partitioning
The partitioning of drugs in microscopic environments (e.g.: lipid bilayers of biological membranes) of different lipophilicity and hydrophilicity heavily influences its pharmacokinetic behavior. The partitioning is described by the logP and logD values of a drug, which are major descriptors in predicting ADMET properties.
The Partitioning bundle provides calculation of:
- LogP values using scientifically sound methods - LogP documentation
- LogD (pH vs. logD) plot - LogD documentation
-
HLB (Hydrophilic Lipophilic Balance) number
Features
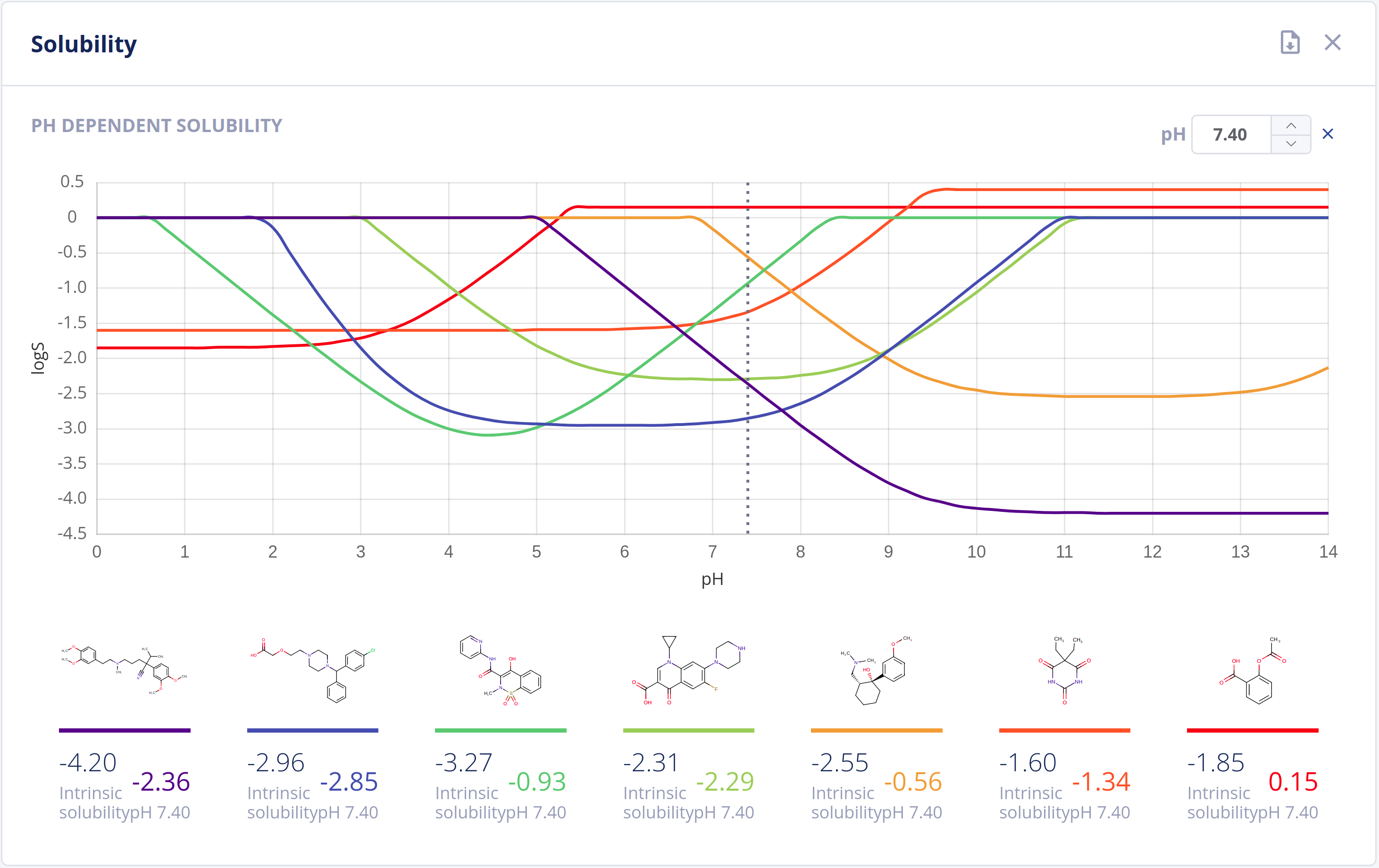
Solubility
Solubility in water (commonly referred to as logS) is one of the most important parameters to achieve for desired pharmacological response. Any drug to be absorbed must be present as a solution at its site of absorption.
As the majority of drugs are molecules that have ionizable groups, the solubility of the compound depends highly on the pH environment. The Solubility Predictor is able to predict:
- Intrinsic (thermodynamic) solubility in water
- pH-dependent solubility (pH-logS plot)
- A qualitative solubility category
Features
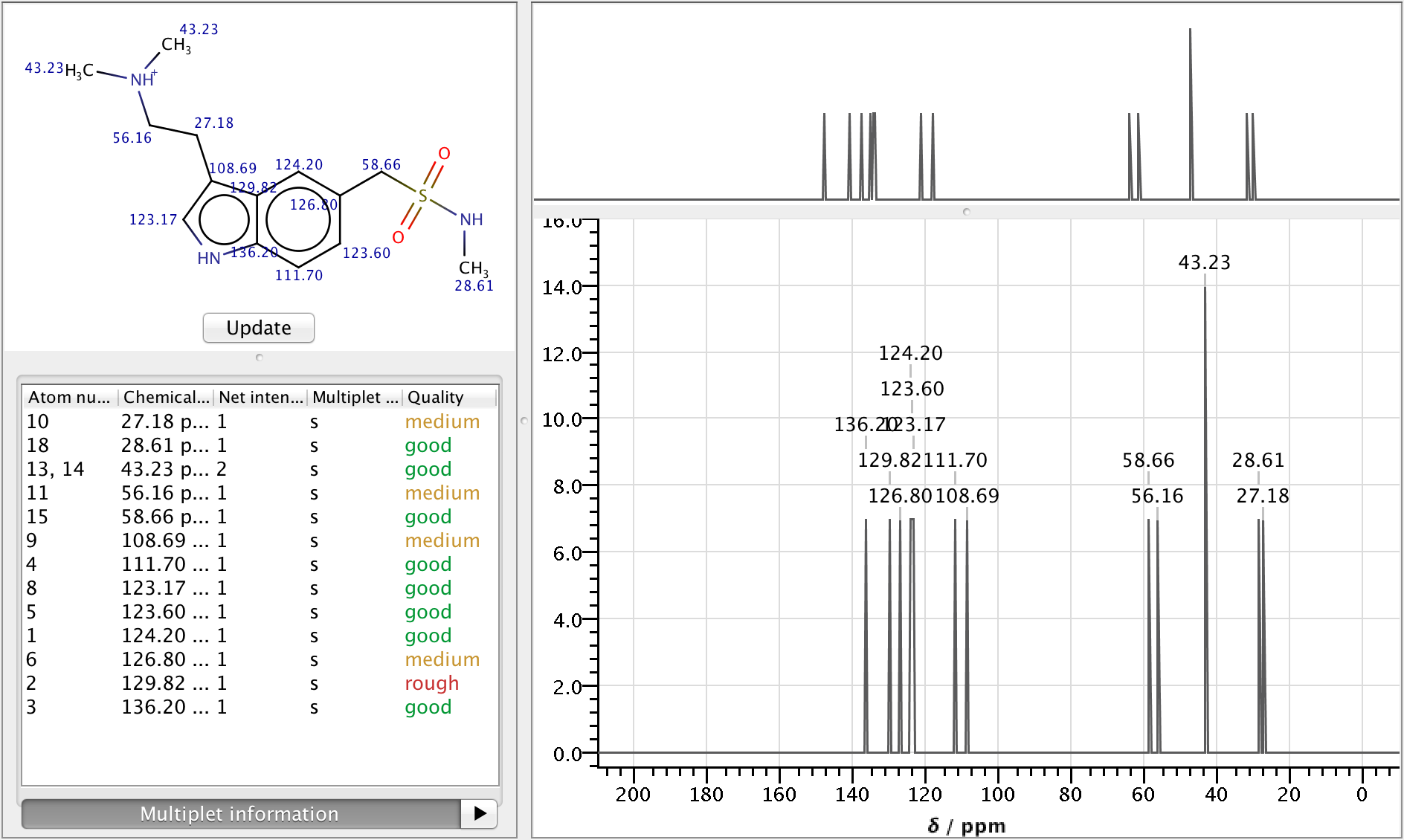
NMR Predictor
Nuclear Magnetic Resonance spectroscopy is an experimental research technique used by scientists to determine chemical structure of molecules.
As NMR spectrometers are relatively expensive, predicting NMR spectra for a set of possible structures and comparing them with experimental data is a well established approach to facilitate structure elucidation. The NMR Predictor is able to:
- Predict 13C and 1H NMR spectra for molecules composed of the most frequent elements (H, C, N, O, F, Cl, Br, I, P, S, Si, Se, B, Sn, Ge, Te and As)
- Import and display NMR spectra from JCAMP-DX files
Features
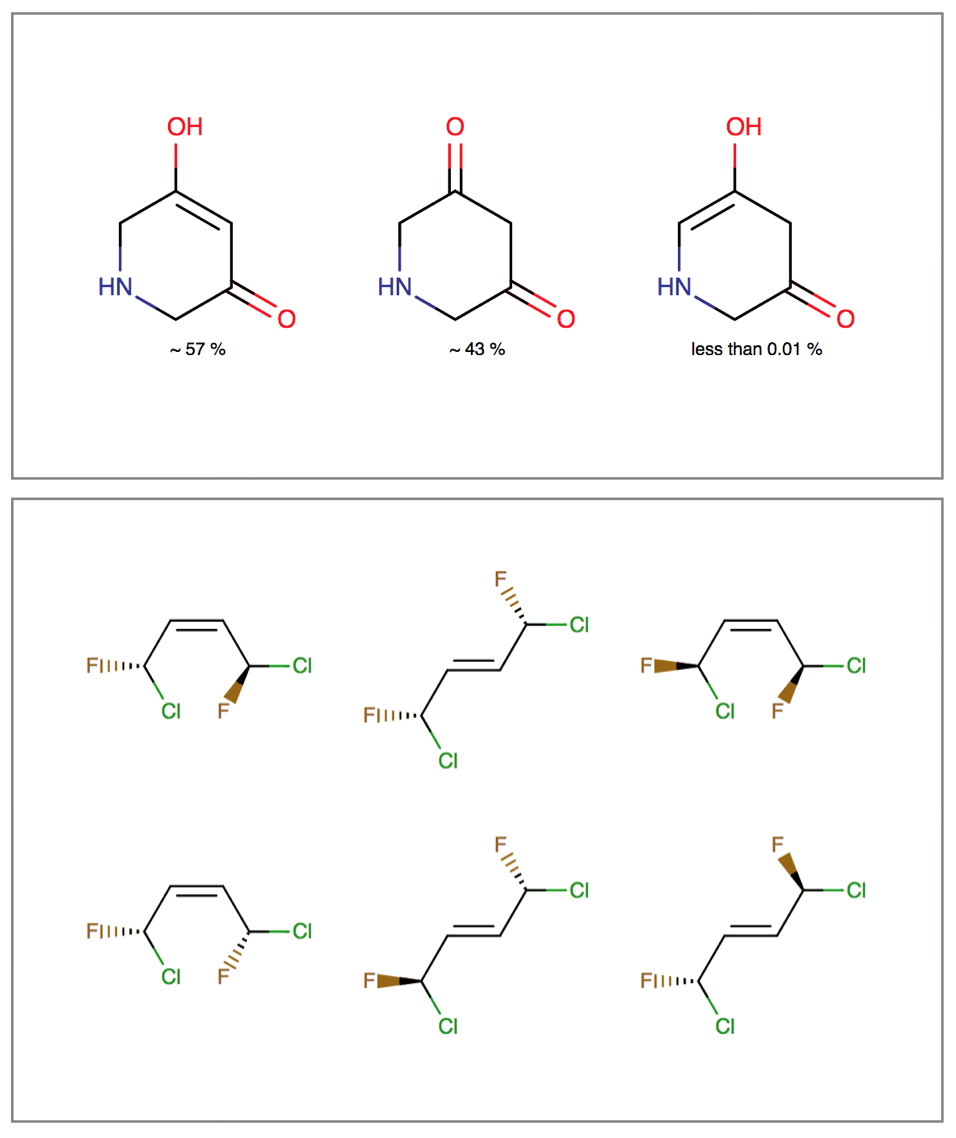
Isomers
Isomerism is an interconversion between molecules that have identical atomic composition, but different arrangement of bonds or spatial orientation. Tautomerism, stereoisomerism and resonance are examples of these inter-conversions.
Tautomerization can affect identification, searching and physico-chemical properties of molecules. Stereoisomers have the same physico-chemical properties (e.g.: melting point, solubility), but they differ in pharmacokinetic and pharmacodynamic behavior. Resonance describes the rearrangement of delocalized electrons in the molecule, which results in a set of contributing structures of the original structure.
The Isomers bundle can:
- Generate different tautomer sets useful for searching and tautomer handling
- Calculate tautomer distribution and major tautomer form in water
- Generate tetrahedral and double-bond stereoisomers starting either from a 2D or 3D structure
- Generate resonance structures
Features
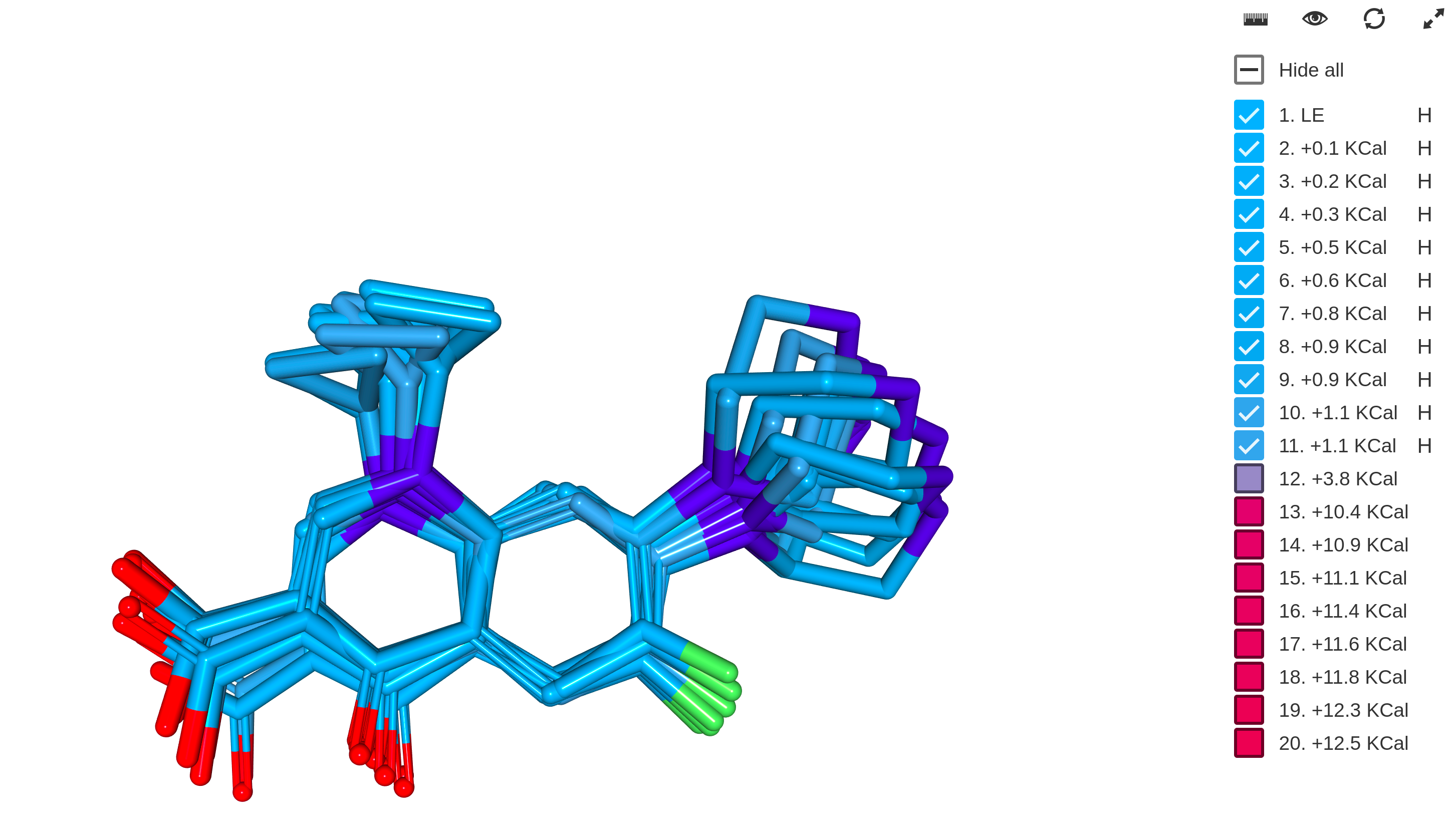
Structural Calculations
The Structural Calculations bundle provides different structural calculations including:
- Hydrogen Bond Donor/Acceptor (HBDA) count
- 2D topological descriptors
- 3D geometrical descriptors
- Molecular surface calculations
- 3D conformer generation, molecular dynamics
- 3D Alignment
- Different electron structural property calculations
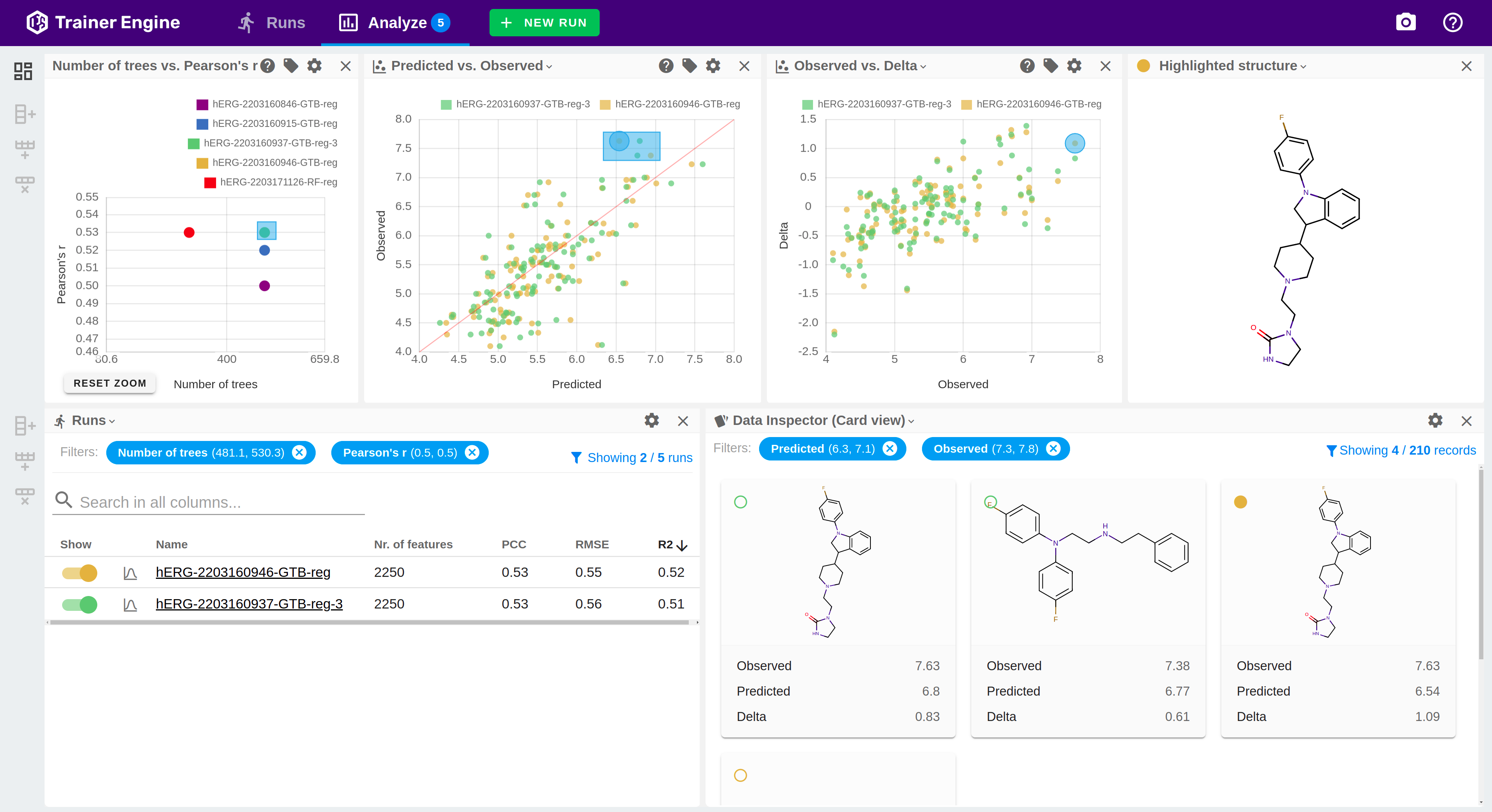
Trainer Engine
Translate data to prediction
Trainer Engine makes chemical, physical and biological activity predictions available by streamlining learning from input data with high accuracy, reliability and confidence at scale. The framework simplifies sharing models, and managing the machine learning lifecycle.
Availability
The versatile Calculators & Predictors technology is available through a number of other applications and can be accessed in multiple ways. Smart interfaces fitting the environment you can work the most effectively:
- Calculator plugins for molecules sketched in Marvin
- Analyzing charts and prediction results via Playground or Chemicalize
- Organizing data, calculation functions in JChem for Office
- Preparing large sets, batch processing via command line tool cxcalc
- Using from other Chemaxon products, Chemical Terms functions are available to access calculations in JChem Engines, Instant JChem, JChem for Office, Compound Registration or Reactor
- Working with workflow tools: calculator nodes in KNIME and components in Pipeline Pilot
- Integrating: Java API, REST API
-
Secure, stateless SaaS REST API for integration: Calculators and Predictors on AWS Marketplace




Subscribe to Chemicalize
Calculate properties instantly, search chemical data, and draw molecules online
Our Chemicalize platform makes Chemaxon technology, used by top pharma companies, easily accessible for both corporate and academic users.

